Written by Daniella H. Hock, Department of Biochemistry and Pharmacology, Bio21 Institute, The University of Melbourne, Australia; David Thorburn, Murdoch Children’s Research Institute, Royal Children’s Hospital, Department of Paediatrics, University of Melbourne, Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Royal Children’s Hospital, Australia; David A. Stroud, Department of Biochemistry and Pharmacology, Bio21 Institute, The University of Melbourne, Australia
Mitochondria are central to energy metabolism, producing the vast majority of cellular ATP via the oxidative phosphorylation (OXPHOS) system, while also being involved in other important processes such as one carbon metabolism, calcium homeostasis, and Fe-S cluster biogenesis (1, 2). Mitochondria are fascinating organelles as they possess their own circular DNA (mtDNA) which encodes 13 proteins translated via their own ribosomes (mitoribosome) while the remaining proteins are translated in the cytosol and imported into the mitochondria (3).
Mitochondrial disease is an umbrella term for a collection of rare genetic disorders where cells fail to produce enough energy for proper organ and tissue function (2, 4). Collectively, it affects at least 1 in 5,000 live births (5) with infants affected by the disease having a particularly poor prognosis. From the over 1,100 known proteins localised to the human mitochondria (6), at least 350 are encoded by genes in which mutations are known to cause mitochondrial disease (4). Mitochondrial disease displays extreme clinical and genetic heterogeneity, can develop at any stage of life, and usually affects organs with high energy demand such as brain, heart, liver and skeletal muscle (2, 4).
Once mitochondrial disease is suspected based on clinical investigation, the search for the genetic variant starts (Fig. 1A). The current diagnostic paradigm typically involves massively parallel sequencing (MPS) approaches such as whole-exome sequencing (WES) and whole-genome sequencing (WGS). Despite these combined approaches, the diagnosis of mitochondrial disease is only achieved in 35-60% of suspected cases and often after years of investigation (7). For some unsolved cases, more than one variant of uncertain significance (VUS) is flagged following bioinformatic analysis, potentially requiring further functional validation to confirm pathogenicity e.g., spectrophotometric assays of OXPHOS enzyme activity and western blot analysis of single proteins. Alternatively a single pathogenic variant is found in a gene associated with autosomal recessive inheritance and it can be unclear if a second “cryptic variant” has been missed. More recently high throughput omic approaches, such as transcriptomics and proteomics, have begun to be integrated into investigations of variant pathogenicity (8). The ‘unbiased’ nature of these techniques also lends themselves to gene discovery in cases where no candidate VUS are identified. The integration of multi-omic approaches into the diagnosis of mitochondrial disease is thus not only decreasing the waiting time for a diagnosis but potentially allowing an earlier use of therapies, disease management and facilitating provision of information that can inform family planning decisions.
Since 2017 we have been developing and applying quantitative proteomics approaches to undiagnosed cases of rare diseases with a focus on patients with clinically suspected mitochondrial disease, while also investigating genes that can potentially cause mitochondrial diseases (9). We have analysed a range of primary tissues, including fibroblasts from skin biopsies, lymphoblasts from blood, myoblasts, skeletal muscle and heart biopsies, and have tested different quantitative approaches to individually address each clinical case. At its most basic, we have applied label free quantitative analysis of proteome changes in whole patient material relative to a panel (usually 3-5) of controls (Fig. 1B). We have also employed fractionation of peptides for increased proteome depth, pulse labelling with stable isotopes to monitor mtDNA translation, isobaric labelling, and other approaches. Our efforts have supported the published (10-13) and unpublished diagnosis of over 30 patients, many of whom were undiagnosed for decades after extensive investigation, thus demonstrating that quantitative proteomics is a technique capable of providing evidence to support disease causation arising from various types of variants, including deep intronic variants, splice site variants, copy number variants, and missense variants present in either mitochondrial or nuclear DNA.
The first cold case we would like to share had been undiagnosed for over 10 years, despite inconclusive results after extensive investigations which included [i] chromosomal microarray and breakage analysis, [ii] quad (both parents and affected siblings) whole exome sequencing, [iii] common mitochondrial DNA point mutations, [iv] and the gold-standard quad whole genome sequencing (11). Transcriptomic analysis (RNA sequencing) was performed and identified a reduction in NDUFB10 expression as well as novel splicing events, including the presence of a cryptic exon in NDUFB10 transcripts (Fig. 1C). We performed label free quantitative proteomics on skin fibroblasts from the patient and three control individuals. NDUFB10 peptides were actually not detected in control or patient cells but we confirmed the reduction in over twenty NDUFB10 partner proteins in patient cells, similar to the protein signature observed in knockout NDUFB10 cells (14). These proteins, along with NDUFB10 are subunits of the first complex in the mitochondrial respiratory chain, also called Complex I or NADH ubiquinone oxidoreductase. The abundance of subunits in the other complexes of the OXPHOS system were unaffected. Reanalysis of WGS data confirmed the presence of a novel deep intronic cryptic splicing variant in NDUFB10, found in homozygosity in the affected siblings and in heterozygosity in the parents.
The second investigation involves the diagnosis of 17 patients where inherited deletions and de novo duplication had occurred in the repetitive ATAD3 locus. Mutations in this locus are now known to be one of the five most common causes of paediatric nuclear-encoded mitochondrial disease (12). The ATAD3 locus is comprised of three highly homologous genes in tandem: ATAD3C, ATAD3B and ATAD3A, an arrangement which is exclusive to hominids. The homology of the region makes it prone to nonallelic homologous recombination (NAHR) events, giving rise to copy number variations (CNVs) such as deletions and duplications. Identification of deletions and duplications events in highly homologous genomic regions can be a challenge as these regions can be refractory to the short read sequencing approaches used in WES and WGS methodologies. To overcome this problem, short read approaches were combined with long-read sequencing, transcriptomics and quantitative proteomics analyses to identify recessive biallelic deletions and dominant de novo duplications in the ATAD3 locus (Fig.1D). At the protein level, the recessive biallelic deletions give rise to a fused ATAD3B/ATAD3A protein, while the dominant de novo duplication give rise to a chimeric ATAD3A/ATAD3C protein.
Quantitative proteomics was shown to provide valuable functional data that can add to the body of evidence needed to confirm pathogenicity of VUS in rare and long-term undiagnosed cases. Our current efforts are now focused on developing a high throughput and robust quantitative proteomics pipeline to be incorporated into routine clinical investigations of patients with suspected but undiagnosed rare disease in order to decrease the current diagnostic gap.
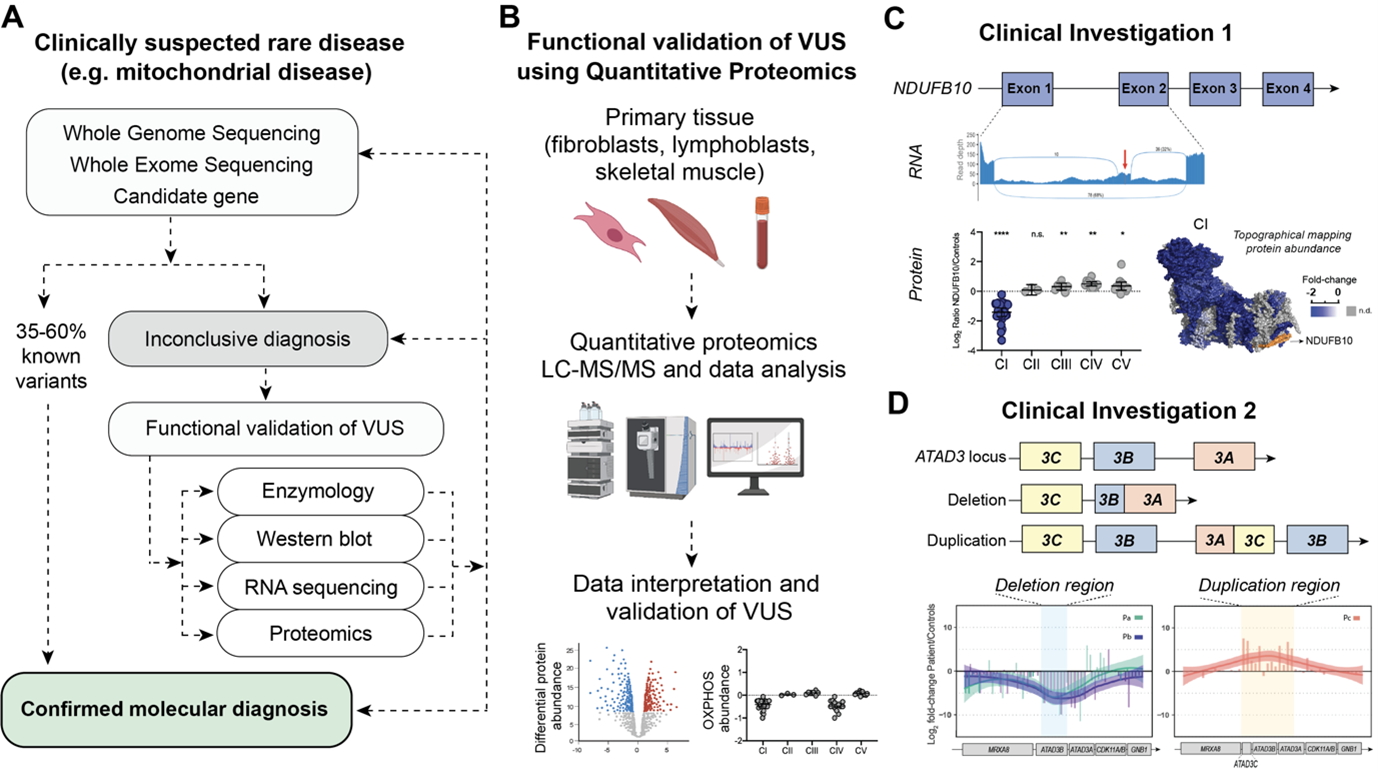
Figure 1A. Example pipeline for investigation of clinically suspected rare disease. Genomic sequencing is the first line approach followed by functional validation of variants of uncertain significance (VUS) using e.g. enzymology, western blot, transcriptomics and proteomics. B. General schematic depicting workflow for functional validation of VUS via quantitative proteomics. C. Multi-omic investigation used to identify a deep intronic variant in NDUFB10, a structural subunit of mitochondrial Complex I (CI). Transcriptomic analysis identified a cryptic exon inserted between exon 1 and exon 2 of NDUFB10. Proteomics on the patient skin fibroblasts show an isolated decrease in CI. Differential protein abundances were topographically mapped to the CI structure (PDB ID: 5LDW) for visualization of the impact of mutation. Data plotted from supplemental material in (11). D. Pathogenic genetic arrangements in the ATAD3 locus identified in patients with a large deletion or duplication in the repetitive ATAD3 locus. Deep peptide fractionation and label free quantitative proteomics of patient skin fibroblasts revealed the corresponding decrease and increase in peptide abundance. Data plotted from supplemental material in (12).
References
1. Jackson TD, Hock DH, Fujihara KM, Palmer CS, Frazier AE, Low YC, et al. The TIM22 complex mediates the import of sideroflexins and is required for efficient mitochondrial one-carbon metabolism. Mol Biol Cell. 2021;32(6):475-91.
2. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem. 2019;294(14):5386-95.
3. Kang Y, Fielden LF, Stojanovski D. Mitochondrial protein transport in health and disease. Semin Cell Dev Biol. 2018;76:142-53.
4. Hock DH, Robinson DRL, Stroud DA. Blackout in the powerhouse: clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem J. 2020;477(21):4085-132.
5. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905-12.
6. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021;49(D1):D1541-D7.
7. Stenton SL, Prokisch H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine. 2020;56:102784.
8. Alston CL, Stenton SL, Hudson G, Prokisch H, Taylor RW. The genetics of mitochondrial disease: dissecting mitochondrial pathology using multi-omic pipelines. J Pathol. 2021.
9. Hock DH, Reljic B, Ang CS, Muellner-Wong L, Mountford HS, Compton AG, et al. HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV. Mol Cell Proteomics. 2020.
10. Lake NJ, Webb BD, Stroud DA, Richman TR, Ruzzenente B, Compton AG, et al. Biallelic Mutations in MRPS34 Lead to Instability of the Small Mitoribosomal Subunit and Leigh Syndrome. Am J Hum Genet. 2017;101(2):239-54.
11. Helman G, Compton AG, Hock DH, Walkiewicz M, Brett GR, Pais L, et al. Multiomic analysis elucidates Complex I deficiency caused by a deep intronic variant in NDUFB10. Hum Mutat. 2021;42(1):19-24.
12. Frazier AE, Compton AG, Kishita Y, Hock DH, Welch AE, Amarasekera SSC, et al. Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the ATAD3 locus. Med (N Y). 2021;2(1):49-73.
13. Van Bergen NJ, Ahmed SM, Collins F, Cowley M, Vetro A, Dale RC, et al. Mutations in the exocyst component EXOC2 cause severe defects in human brain development. J Exp Med. 2020;217(10).
14. Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature. 2016;538(7623):123-6.
Biography

Daniella Hock is a PhD candidate in Dr David Stroud’s lab at the University of Melbourne in Australia, where she has been developing and applying quantitative proteomics approaches to improve the diagnostic rates of rare diseases, particularly paediatric mitochondrial disease. She is also interested in investigating the molecular function of genes implicated in mitochondrial disease pathogenesis with a view toward expanding the list of genes that can potentially cause mitochondrial disease. Prior to commencing her PhD, Daniella graduated as Bachelor of Biological Sciences from Universidade Federal de Santa Catarina in Brazil and worked as molecular diagnostic scientist at Biogenetika, a personalised medicine centre, in Brazil.


.png)












